Detailed walk-through of de novo CIDER (dnCIDER) on pancreas data
Source:vignettes/dnCIDER.Rmd
dnCIDER.RmdIntroduction
This vignette performs dnCIDER on a cross-species pancreas dataset. It is aimed to show the underneath structure of dnCIDER compared to the other high-level vignette (see Getting Start with De Novo CIDER (dnCIDER): Cross-Species Pancreas Integration).
Set up
In addition to CIDER, we will load the following packages:
library(CIDER)
library(Seurat)
#> Loading required package: SeuratObject
#> Loading required package: sp
#> 'SeuratObject' was built under R 4.4.0 but the current version is
#> 4.4.1; it is recomended that you reinstall 'SeuratObject' as the ABI
#> for R may have changed
#>
#> Attaching package: 'SeuratObject'
#> The following objects are masked from 'package:base':
#>
#> intersect, t
library(parallel)
library(cowplot)Load example data
The example data can be downloaded from https://figshare.com/s/d5474749ca8c711cc205.
Pancreatic cell data contain cells from human (8241 cells) and mouse (1886 cells).
# Download the data
data_url <- "https://figshare.com/ndownloader/files/31469387"
data_file <- file.path(tempdir(), "pancreas_counts.RData")
if (!file.exists(data_file)) {
message("Downloading data...")
download.file(data_url, destfile = data_file, mode = "wb")
}
#> Downloading data...
data_env <- new.env()
loaded_objects <- load(file = data_file, envir = data_env)
pancreas_counts <- data_env[[loaded_objects[1]]]
load("../data/pancreas_meta.RData") # meta data/cell information
seu <- CreateSeuratObject(counts = pancreas_counts, meta.data = pancreas_meta)
table(seu$Batch)
#>
#> human mouse
#> 8241 1886Perform initial clustering
seu_list <- Seurat::SplitObject(seu, split.by = "Batch")
seu_list <- mclapply(seu_list, function(x) {
x <- NormalizeData(x, normalization.method = "LogNormalize",
scale.factor = 10000, verbose = FALSE)
x <- FindVariableFeatures(x, selection.method = "vst",
nfeatures = 2000, verbose = FALSE)
x <- ScaleData(x, verbose = FALSE, vars.to.regress = "Sample")
x <- RunPCA(x, features = VariableFeatures(object = x), verbose = FALSE)
x <- FindNeighbors(x, dims = 1:15, verbose = FALSE)
x <- FindClusters(x, resolution = 0.6, verbose = FALSE)
return(x)
})| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| acinar | 0 | 0 | 823 | 0 | 0 | 0 | 1 | 1 | 2 | 102 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 |
| activated_stellate | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 269 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 |
| alpha | 1067 | 3 | 0 | 0 | 0 | 665 | 0 | 0 | 0 | 0 | 251 | 243 | 1 | 6 | 0 | 0 | 1 | 0 | 4 |
| b_cell | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| beta | 1 | 886 | 2 | 812 | 740 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 3 | 2 | 1 | 0 | 4 | 0 | 1 |
| delta | 0 | 3 | 2 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 215 | 1 | 216 | 0 | 148 | 0 | 2 |
| ductal | 1 | 1 | 6 | 0 | 0 | 0 | 469 | 386 | 0 | 169 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| endothelial | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 212 | 0 | 1 | 0 |
| epsilon | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 8 | 0 | 0 | 1 | 0 | 0 |
| gamma | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 3 | 210 | 0 | 0 | 25 | 0 | 0 |
| immune_other | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| macrophage | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 39 |
| mast | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 25 |
| quiescent_stellate | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 153 | 0 |
| schwann | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| t_cell | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 |
seu_list <- mclapply(seu_list, RunTSNE, dims = 1:15)
p1 <- scatterPlot(seu_list[[1]], "tsne", colour.by = "seurat_clusters")
p2 <- scatterPlot(seu_list[[2]], "tsne", colour.by = "seurat_clusters")
cowplot::plot_grid(p1,p2)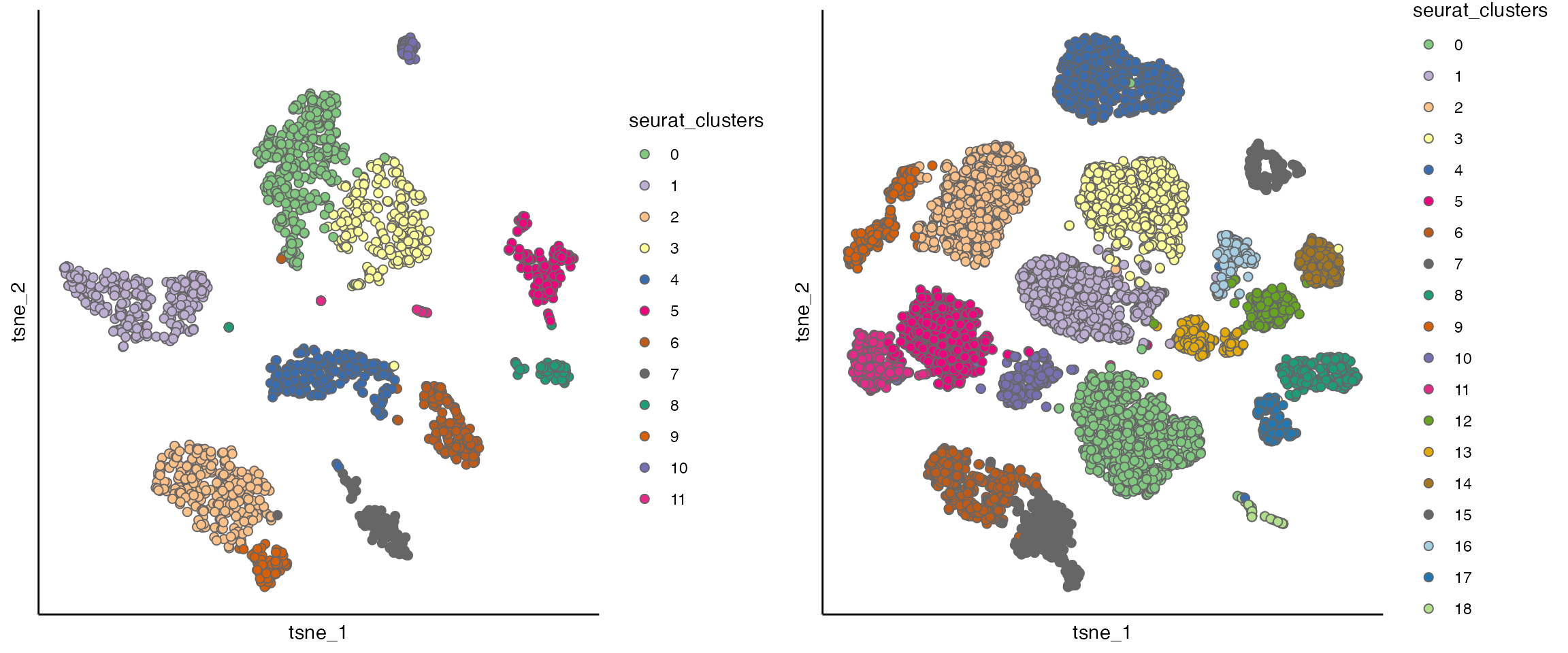
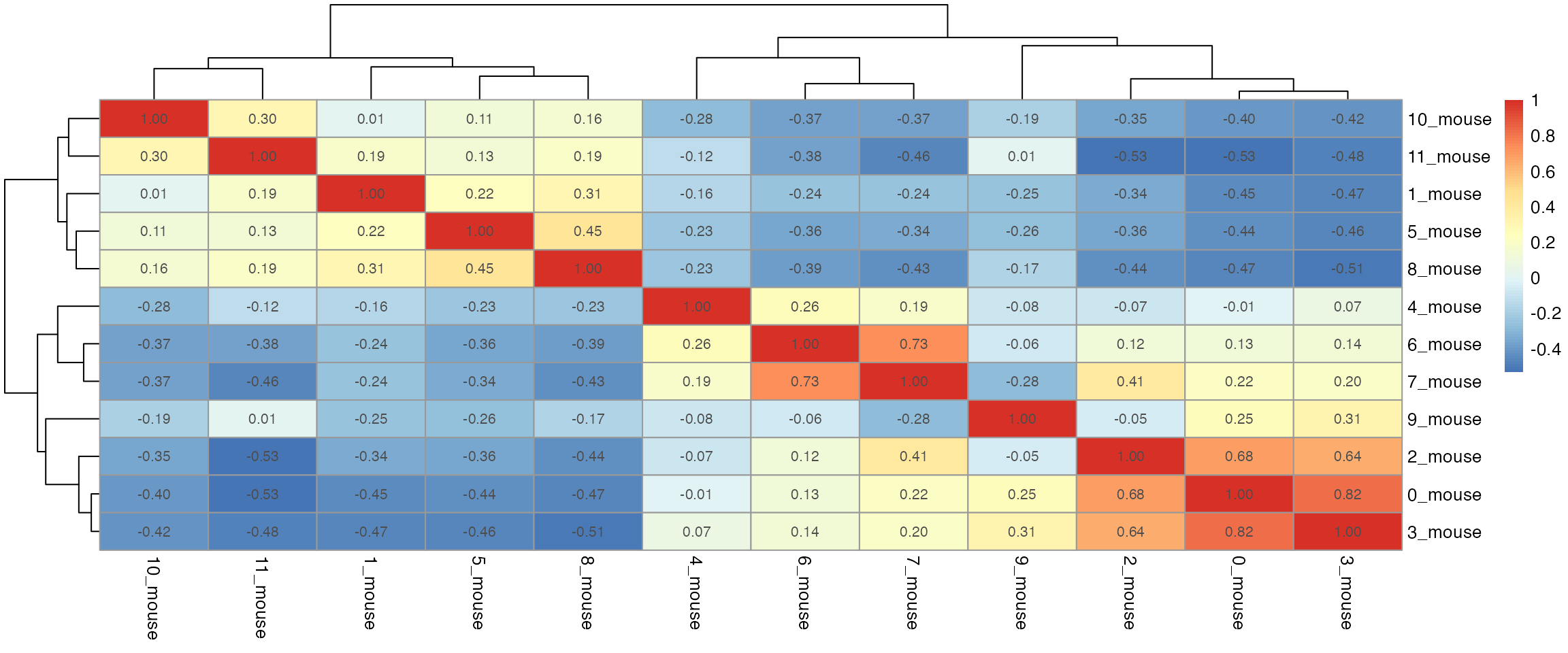
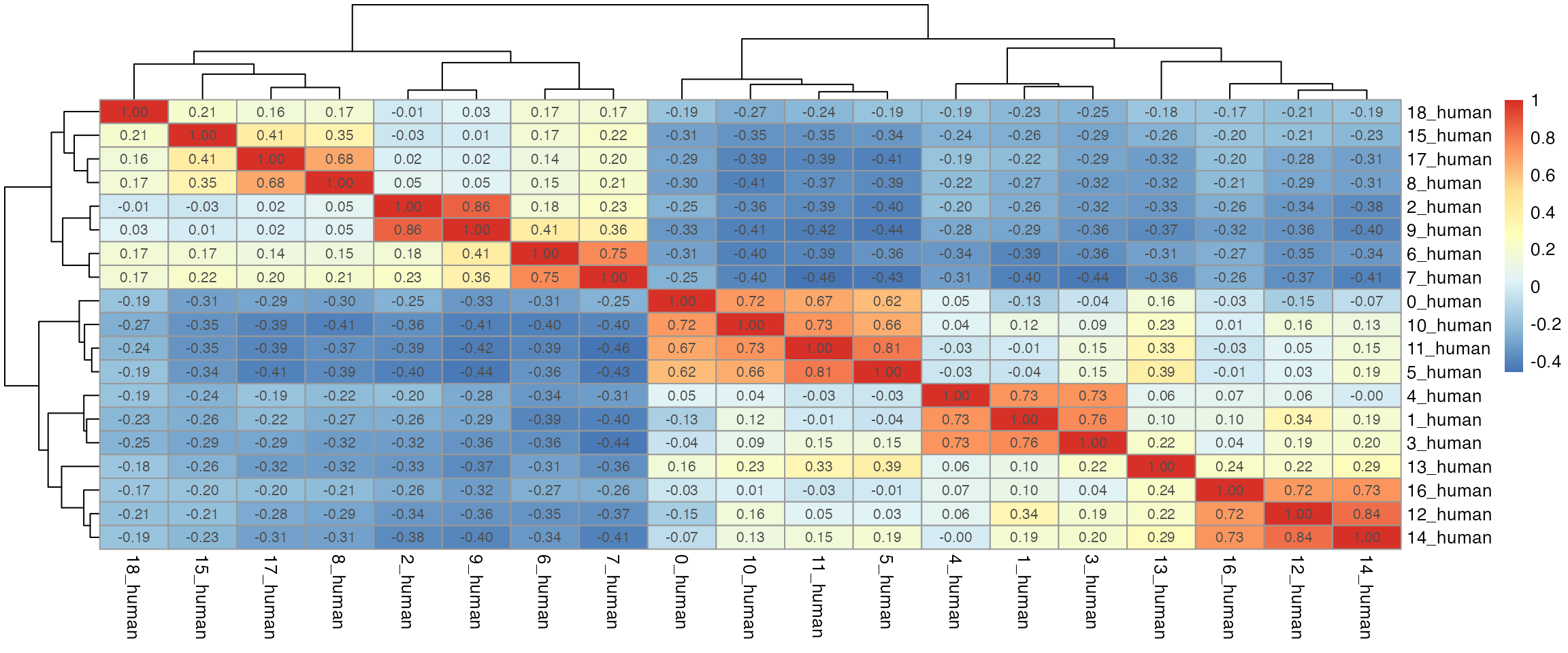
dist_coef <- getDistMat(seu_list, downsampling.size = 50)
#> | | | 0% | |=================================== | 50% | |======================================================================| 100%
par(mfrow = c(length(seu_list),1))
for(i in which(sapply(dist_coef, function(x) return(!is.null(x))))){
tmp <- dist_coef[[i]] + t(dist_coef[[i]])
diag(tmp) <- 1
pheatmap::pheatmap(tmp, display_numbers = TRUE)
}
for(seu_itor in 1:2){
tmp <- dist_coef[[seu_itor]] + t(dist_coef[[seu_itor]])
diag(tmp) <- 1
tmp <- 1 - tmp
hc <- hclust(as.dist(tmp), method = "average")
hres <- cutree(hc, h = 0.4)
df_hres <- data.frame(hres)
df_hres$hres <- paste0(df_hres$hres, "_", unique(seu_list[[seu_itor]]$Batch))
seu_list[[seu_itor]]$inicluster_tmp <- paste0(seu_list[[seu_itor]]$seurat_clusters, "_", seu_list[[seu_itor]]$Batch)
seu_list[[seu_itor]]$inicluster <- df_hres$hres[match(seu_list[[seu_itor]]$inicluster_tmp,rownames(df_hres))]
}
# plot(as.dendrogram(hc), horiz = T)
p1 <- scatterPlot(seu_list[[1]], "tsne", "inicluster")
p2 <- scatterPlot(seu_list[[2]], "tsne", "inicluster")
plot_grid(p1,p2)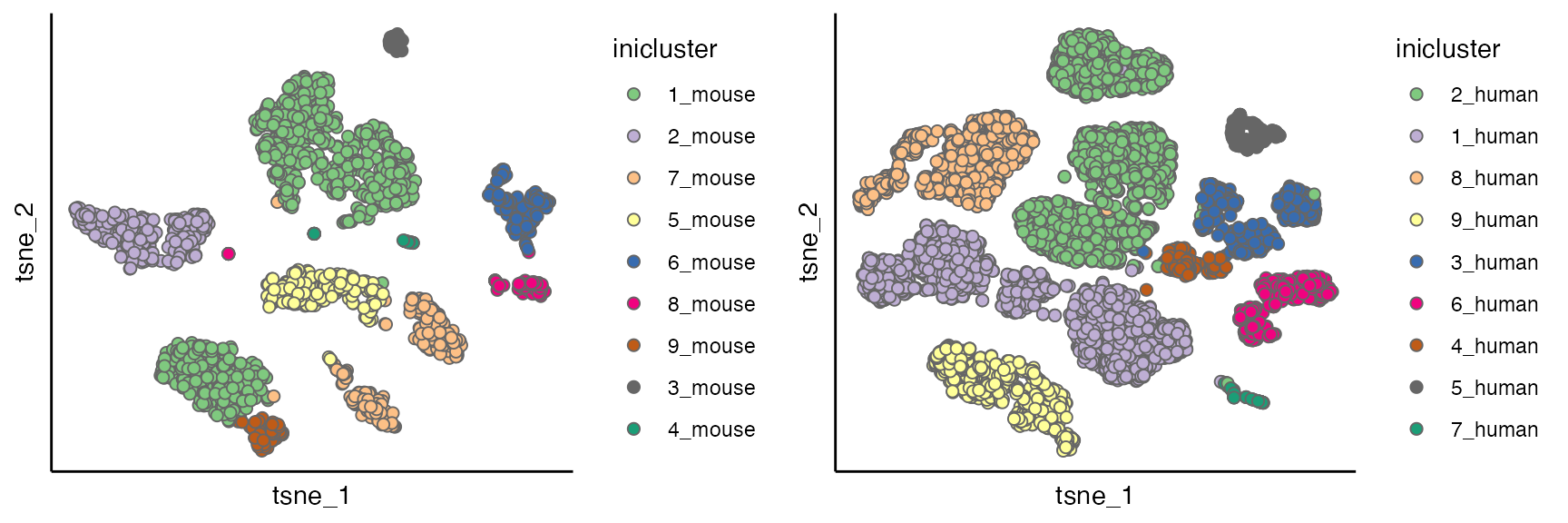
scatterPlot(seu_list[[2]], "tsne", "Group")
Calculate of IDER similarity matrix
res <- unlist(lapply(seu_list, function(x) return(x$inicluster)))
res_names <- unlist(lapply(seu_list, function(x) return(colnames(x))))
seu@meta.data$initial_cluster <- res[match(colnames(seu), res_names)]
ider <- getIDEr(seu,
group.by.var = "initial_cluster",
batch.by.var = "Batch",
downsampling.size = 35,
use.parallel = FALSE, verbose = FALSE)
net <- plotNetwork(seu, ider, colour.by = "Group" , vertex.size = 0.6, weight.factor = 5)
hc <- hclust(as.dist(1-(ider[[1]] + t(ider[[1]])))/2)
plot(as.dendrogram(hc)) #, horiz = TRUE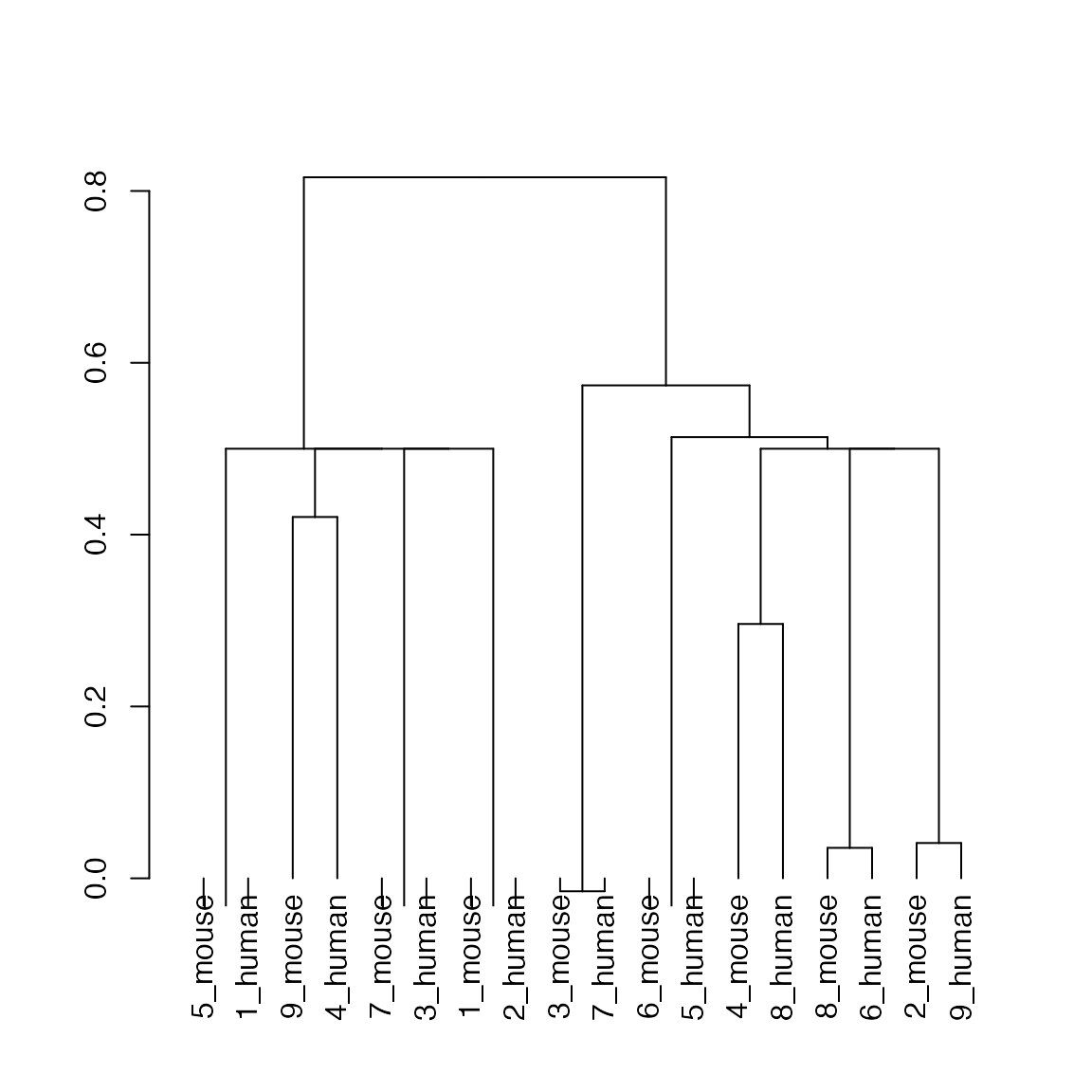
Perform final Clustering
seu <- finalClustering(seu, ider, cutree.h = 0.35) # final clustering
seu <- NormalizeData(seu, verbose = FALSE)
seu <- FindVariableFeatures(seu, selection.method = "vst",
nfeatures = 2000, verbose = FALSE)
seu <- ScaleData(seu, verbose = FALSE)
seu <- RunPCA(seu, npcs = 20, verbose = FALSE)
seu <- RunTSNE(seu, reduction = "pca", dims = 1:12)
plot_list <- list()
plot_list[[1]] <- scatterPlot(seu, "tsne", colour.by = "CIDER_cluster", title = "asCIDER clustering results")
plot_list[[2]] <- scatterPlot(seu, "tsne", colour.by = "Group", title = "Ground truth of cell populations")
plot_grid(plotlist = plot_list, ncol = 2)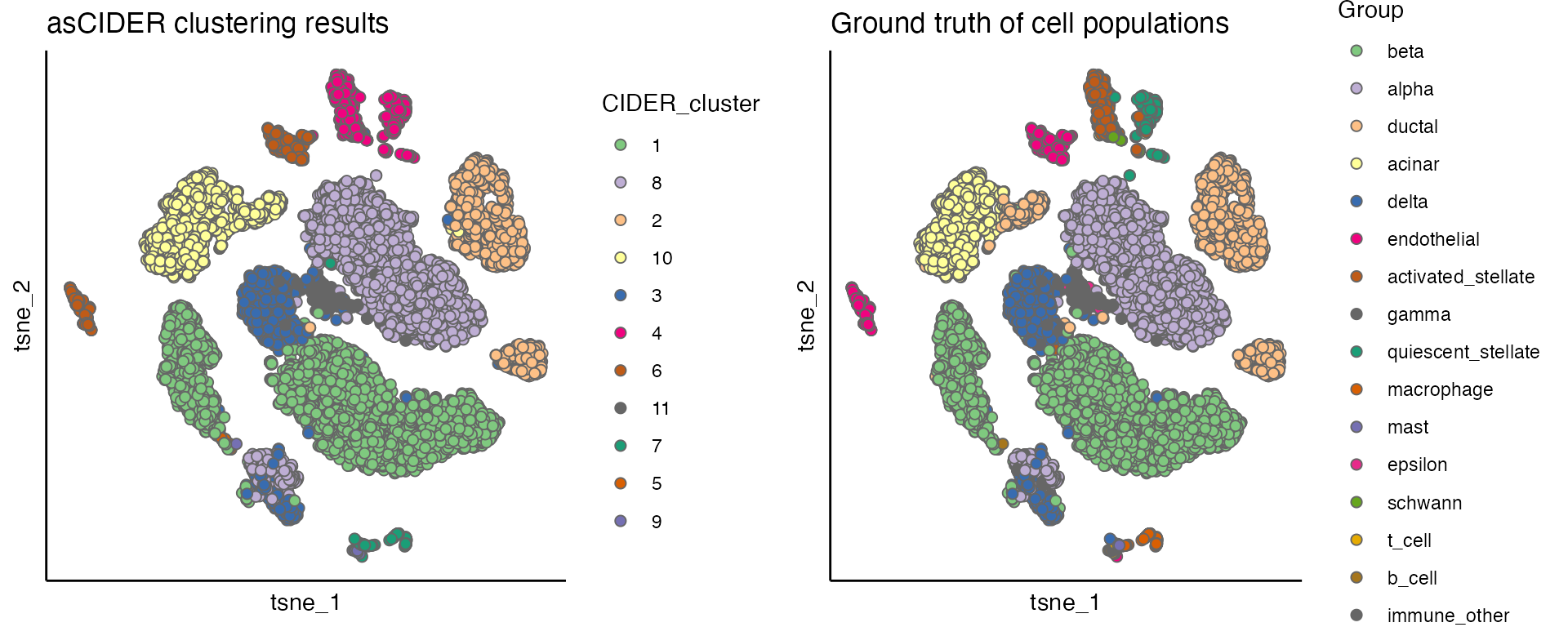
Reproducibility
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: x86_64-apple-darwin20
#> Running under: macOS Monterey 12.5.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Europe/London
#> tzcode source: internal
#>
#> attached base packages:
#> [1] parallel stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] cowplot_1.1.3 Seurat_5.1.0 SeuratObject_5.0.2 sp_2.1-4
#> [5] CIDER_0.99.4
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 rstudioapi_0.16.0 jsonlite_1.8.8
#> [4] magrittr_2.0.3 spatstat.utils_3.1-0 farver_2.1.2
#> [7] rmarkdown_2.27 fs_1.6.4 ragg_1.3.2
#> [10] vctrs_0.6.5 ROCR_1.0-11 spatstat.explore_3.3-2
#> [13] htmltools_0.5.8.1 sass_0.4.9 sctransform_0.4.1
#> [16] parallelly_1.38.0 KernSmooth_2.23-24 bslib_0.7.0
#> [19] htmlwidgets_1.6.4 desc_1.4.3 ica_1.0-3
#> [22] plyr_1.8.9 plotly_4.10.4 zoo_1.8-12
#> [25] cachem_1.1.0 igraph_2.0.3 mime_0.12
#> [28] lifecycle_1.0.4 iterators_1.0.14 pkgconfig_2.0.3
#> [31] Matrix_1.7-0 R6_2.5.1 fastmap_1.2.0
#> [34] fitdistrplus_1.2-1 future_1.34.0 shiny_1.9.1
#> [37] digest_0.6.37 colorspace_2.1-1 patchwork_1.2.0
#> [40] tensor_1.5 RSpectra_0.16-2 irlba_2.3.5.1
#> [43] textshaping_0.4.0 labeling_0.4.3 progressr_0.14.0
#> [46] fansi_1.0.6 spatstat.sparse_3.1-0 httr_1.4.7
#> [49] polyclip_1.10-7 abind_1.4-5 compiler_4.4.1
#> [52] withr_3.0.1 doParallel_1.0.17 viridis_0.6.5
#> [55] fastDummies_1.7.4 highr_0.11 MASS_7.3-61
#> [58] tools_4.4.1 lmtest_0.9-40 httpuv_1.6.15
#> [61] future.apply_1.11.2 goftest_1.2-3 glue_1.7.0
#> [64] dbscan_1.2.2 nlme_3.1-165 promises_1.3.0
#> [67] grid_4.4.1 Rtsne_0.17 cluster_2.1.6
#> [70] reshape2_1.4.4 generics_0.1.3 gtable_0.3.5
#> [73] spatstat.data_3.1-2 tidyr_1.3.1 data.table_1.16.0
#> [76] utf8_1.2.4 spatstat.geom_3.3-2 RcppAnnoy_0.0.22
#> [79] ggrepel_0.9.5 RANN_2.6.2 foreach_1.5.2
#> [82] pillar_1.9.0 stringr_1.5.1 limma_3.60.6
#> [85] spam_2.10-0 RcppHNSW_0.6.0 later_1.3.2
#> [88] splines_4.4.1 dplyr_1.1.4 lattice_0.22-6
#> [91] survival_3.7-0 deldir_2.0-4 tidyselect_1.2.1
#> [94] locfit_1.5-9.10 miniUI_0.1.1.1 pbapply_1.7-2
#> [97] knitr_1.48 gridExtra_2.3 edgeR_4.2.2
#> [100] scattermore_1.2 xfun_0.46 statmod_1.5.0
#> [103] matrixStats_1.4.1 pheatmap_1.0.12 stringi_1.8.4
#> [106] lazyeval_0.2.2 yaml_2.3.10 evaluate_0.24.0
#> [109] codetools_0.2-20 kernlab_0.9-33 tibble_3.2.1
#> [112] cli_3.6.3 uwot_0.2.2 xtable_1.8-4
#> [115] reticulate_1.39.0 systemfonts_1.1.0 munsell_0.5.1
#> [118] jquerylib_0.1.4 Rcpp_1.0.13 globals_0.16.3
#> [121] spatstat.random_3.3-1 png_0.1-8 spatstat.univar_3.0-1
#> [124] pkgdown_2.1.0 ggplot2_3.5.1 dotCall64_1.1-1
#> [127] listenv_0.9.1 viridisLite_0.4.2 scales_1.3.0
#> [130] ggridges_0.5.6 leiden_0.4.3.1 purrr_1.0.2
#> [133] rlang_1.1.4References
- Baron, M. et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst 3, 346–360.e4 (2016).
- Satija R, et al. Spatial reconstruction of single-cell gene expression data. Nature Biotechnology 33, 495-502 (2015).
- The count matrix and sample information were downloaded from NCBI GEO accession GSE84133.